








公众号/
编辑 | 萝卜皮
深度学习已经能够成功预测 DNA 序列的表观基因组图谱。大多数方法将此任务定义为二元分类,依赖峰值调用者来定义功能活动。最近,出现了定量模型来直接预测实验覆盖值作为回归。随着具有不同架构和训练配置的新模型不断出现,由于缺乏公平评估所提出模型的新颖性,及其对下游生物发现的效用的能力,一个巨大的瓶颈正在形成。
在这里,美国冷泉港实验室(The Cold Spring Harbor Laboratory)的研究人员介绍了一个统一的评估框架,并用它来比较经过训练以预测染色质可及性数据的各种二元和定量模型。他们重点介绍了影响泛化性能的各种建模选择,包括预测变量效应的下游应用。此外,研究人员引入了一个稳健性指标,可用于增强模型选择和改进变异效应预测。
该研究以「Evaluating deep learning for predicting epigenomic profiles」为题,于 2022 年 12 月 5 日发布在《Nature Machine Intelligence》。

深度学习(DL)在预测 DNA 序列的表观基因组图谱方面取得了相当大的成功,包括转录因子结合、染色质可及性、甲基化和组蛋白标记。通过学习序列-功能关系,经过训练的 DL 模型已被用于各种下游任务,例如预测与人类疾病相关的单核苷酸变异的功能影响。
在过去的几年中,为解决调控基因组预测任务而提出的各种 DL 模型已大大增加。各种各样的提议模型、训练它们的数据集、数据集的处理方式以及用于训练模型的技巧,使得评估哪些创新正在推动性能提升变得具有挑战性。由于预测任务的框架方式各不相同,因此并不总是很容易直接比较模型性能。例如,以前的方法通常将任务构建为二元分类,其中二元标签表示基于峰值呼叫者的功能活动。
然而,在将峰的振幅和形状折叠成二进制标签时,有关可能编码在这些属性中的差异顺式调节机制的信息丢失了。最近,出现了定量模型,同样将 DNA 序列作为输入,但现在直接预测实验读取覆盖值作为回归任务,从而绕过了对峰值调用者的需要并保留了表观基因组轨迹的定量信息。由于分类和回归任务的标准指标不同,因此目前还不清楚如何直接比较在二元任务和定量任务上训练的模型。
为了解决这个问题,Kelley 团队建议使用峰值识别器对他们的定量预测进行「二值化」,这样可以将重叠区域与二进制标签进行比较。然而,这种方法将评估狭隘地集中在根据峰值调用者注释为功能的基因组区域,该区域有噪声并且对峰值调用者的参数选择敏感。或者,Avsec 团队将二元模型的性能与二元模型的增强版本进行了比较,后者附加了一个同时预测定量配置文件的输出头。虽然这衡量了定量建模的额外好处,但这种方法需要重新训练模型的多个版本,这可能对初始化敏感,并且不容易扩展到与现有模型的比较。
此外,预测任务中的其他建模选择使得直接进行公平比较具有挑战性。例如,现有的定量模型预测表观遗传图谱的不同分辨率。Basenji 以 128 个碱基对 (bp) 的分辨率预测非重叠分箱表观基因组概况,而 BPNet 以碱基分辨率预测。比较不同分辨率的模型并不简单,因为合并会影响覆盖值的平滑度,进而影响性能指标。此外,现有方法采用不同的数据扩充并分析训练和测试数据的不同子集,进一步使任何直接比较复杂化。
随着应用程序数量的不断增长,建模创新的瓶颈正在形成,因为科学家缺乏对新提出的模型进行批判性评估的能力。
在这里,冷泉港实验室的研究人员提出了一个针对监管基因组学数据训练的 DL 模型的评估框架,称为 GOPHER(GenOmic Profile-model compreHensive EvaluatoR)。无论预测任务的框架如何,该框架都可以对预测性能和模型可解释性进行系统比较。

图:GOPHER 概述。(来源:论文)
使用此框架,研究人员对染色质可及性预测任务的定量模型和二元模型进行了严格评估,以阐明模型架构、训练过程和数据增强策略中的有益因素。超越预测性能,研究人员使用额外的标准评估每个模型:(1)预测对输入序列的小扰动的稳健性,(2)变异效应预测,(3)学习表示的可解释性。
虽然之前的软件,如 Janggu 和 Selene,有助于处理生物数据(主要集中在以峰值为中心的数据)和高级 API 分别在 TensorFlow 和 Pytorch 中训练神经网络模型,它们不关注跨不同预测任务的下游评估。
相比之下,GOPHER 提供了一个全面的模型评估框架,除了使用各种数据扩充训练自定义深度学习模型外,还支持基于峰值的二元分类和 bigWig 轨迹的定量回归分析的数据处理。GOPHER 还结合了许多流行的模型可解释性工具,例如第一层过滤器可视化、全局重要性分析和归因方法,包括计算机诱变、显著图、集成梯度和 SmoothGrad。
使用 GOPHER,研究人员解决了几个悬而未决的问题:(1)如何公平地比较二元模型和定量模型;(2)损失函数的选择如何影响性能;(3)数据集选择如何影响模型性能;(4)如何比较在不同分辨率下进行预测的定量模型;(5)增强策略如何影响模型性能和对平移扰动的稳健性;建模选择如何影响下游(6)功能变异效应预测和(7)模型可解释性。
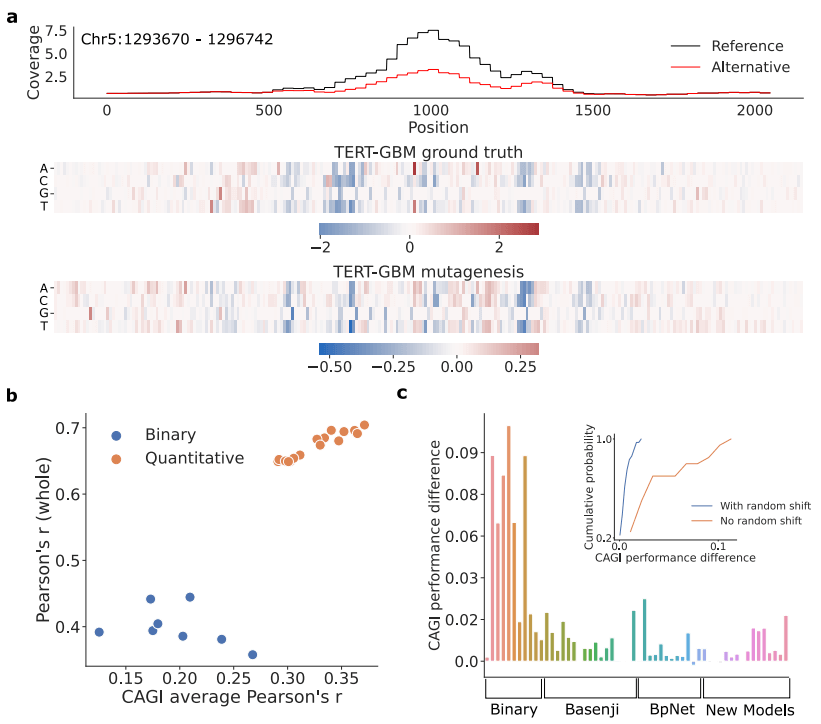
图:功能效应预测的比较。(来源:论文)
虽然此处的研究侧重于 ATAC-seq 数据,但最佳架构和训练程序的具体声明可能在其他数据类型(例如 ChIP-nexus 和 CAGE-seq)中存在细微差别。在这种情况下,可能会出现其他考虑因素,例如 GC 偏差和信号归一化。研究人员表示这些方面还有待进一步研究。
另外,该研究中探索的许多架构都依赖于纯卷积网络。Transformer 的出现可能具有更好的归纳偏差来捕获远端交互,尽管基于卷积的网络和基于 Transformer 的网络的好处的基本原理仍然是一个正在进行的研究课题。由于在 Enformer 之外缺乏已建立的基于 Transformer 的模型,该研究中仅关注基于卷积的模型。
此外,BPNet 和 Basenji 最初是为预测非常不同的数据类型和数据集大小而开发的。因此,必须对每个模型架构进行修改,以使其适应本研究中使用的 ATAC-seq 数据。这些选择可能影响了它们的表现。两个模型都表现良好的事实表明这里的的超参数优化减轻了任何实质性差异。
总的来说,该团队的工作在很大程度上支持定量建模在保留数据和 OOD 变量效应预测方面产生更好的泛化(平均)。当然,调整良好的二元模型可以与设计不佳的定量模型相媲美(甚至更好)。
目前尚不清楚二元模型是否从根本上限制了它们对功能活动的处理,或者在训练过程中加入更多的非活动区域是否会提高性能。
此外,尚不清楚定量模型的性能提升是由于学习了更好的生物信号,还是它们只是更好地学习了测序实验中的噪声源。一个主要的局限性是由于关注性能而产生的——将实验测量视为基本事实,尽管重复和技术噪音之间存在生物学差异。因此,关注重要的下游任务,例如变体效应预测和模型可解释性,就像这里所做的那样,提供了一条超越性能基准的途径,可以转向基因组 DL 模型的有益用例——生物发现。
论文链接:https://www.nature.com/articles/s42256-022-00570-9
相关报道:https://techxplore.com/news/2022-12-ai.html

 友情链接
友情链接