








公众号/
编辑 | 绿萝

人工智能正在改变现代电子产品——加速可弯曲电视屏幕、超轻型革命性太阳能电池等的设计。
具有定制特性的分子和材料的设计具有挑战性,因为候选分子必须满足通常难以测量或计算的多种竞争要求。虽然通过生成式深度学习产生的分子结构将满足这些模式,但它们通常只是偶然而非设计地拥有特定的目标属性,这使得通过这种途径进行分子发现效率低下。
近日,来自英国华威大学(University of Warwick)和德国莱比锡大学(Universität Leipzig)的研究人员使用一种人工智能算法来创造新分子。该算法能够通过在计算机上逐个原子地构建,来快速设计出数百万个以前未发现的分子。
研究人员通过将预测分子三维构象的生成式深度学习模型与将这些作为输入并预测其电子结构的监督深度学习模型相结合来预测具有(帕累托)最优特性的分子。(多个)分子特性的优化是通过筛选新生成的分子以获得所需的电子特性并重新使用命中分子来重新训练具有偏差的生成模型来实现的。
该方法被证明可以找到有机电子应用的最佳分子。所提方法具有普遍适用性,无需在预测过程中进行量子化学计算,适用于材料和催化剂设计中的高通量筛选。
该研究以「High-throughput property-driven generative design of functional organic molecules」为题,于 2023 年 2 月 6 日发布在《Nature Computational Science》上。

论文链接:https://doi.org/10.1038/s43588-022-00391-1
设计特定属性的分子和材料有挑战性
寻找新的功能分子和材料,通常会因必须同时满足多个标准而变得复杂。同时多属性优化可以被认为是分子和材料设计中的圣杯。
更好地了解分子中的官能团如何改变其物理化学性质,至少在原则上有助于促进设计研究。然而,由多达 10^60 个有机分子组成的化学空间的组合复杂性和许多必须考虑的许多因素,往往使这个问题对于传统优化和基本启发式推理来说过于复杂。在开发具有特定属性要求的新分子和材料时,基于简单结构-属性关系和试错优化的候选者识别仍然是最先进的。
有机光电子学是研究发光或探测光的设备的一个研究领域。新型有机电子材料发挥作用的例子包括可持续能源(太阳能电池)、有机发光二极管、电信、智能设备中的显示器和光纤等。
有机薄膜器件,由多个有机层组成,具有不同的定制性质。为薄膜器件提供新的分子材料,它们的电子性质,如基本间隙(ΔE)、电子亲和势(EA)或电离势(IP),必须在一个狭窄的窗口内,以满足设备功能的要求。
生成式深度学习发现分子效率低
最近,生成式深度学习(generative deep learning)已成为加速分子设计的一个有前途的解决方案。生成式深度学习是一种无监督学习技术,其中深度学习模型从(分子)几何数据集中提取知识,并应用获得的规则来创建具有与原始数据集中的属性相似的新分子。最近的几项工作表明,此类方法有可能显着加速分子和材料的发现,然而,不能保证生成的分子系统将在相关体制内表现出特性。
化学空间中的无引导搜索效率极低,从根本上限制了可以在高通量筛选中探索的结构的多样性,特别是如果分子生成过程需要计算要求很高的电子特性的量子化学预测。即使假设拥有无限的计算资源,生成分子的表征仍然具有挑战性。
功能有机分子的高通量引导搜索和设计
在这项工作中,研究人员提出了一种方法,该方法可提供具有定制特性的功能性有机分子的高通量引导搜索和设计。
该方法通过结合两种机器学习算法来实现这一点。第一个模型是一个无监督的生成自回归模型,它可以使用从分子结构分布中学习的化学规则来创建新的、以前未知的分子三维平衡构象。第二个模型是受监督的物理启发的深度神经网络,给定三维结构,可以预测功能有机分子的(带电)电子激发,接近实验精度。后者消除了以前方法中使用的要求苛刻的量子化学计算的需要。
此处介绍的方法提供了一个自动化的工作流程,在该工作流程中,化学空间探索可以偏向于生成满足预设设计参数的分子。
工作流程
所提出的自动化分子设计方法是两种深度学习技术的组合,如图 1a 所示。
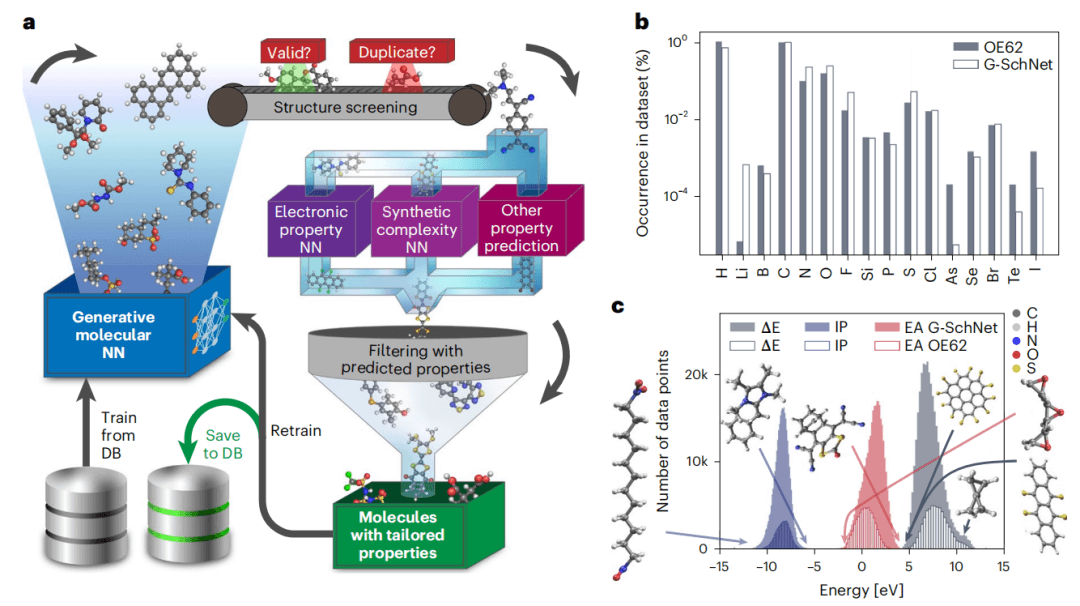
图 1:所提出方法的工作流程和数据集中分子的分布。(来源:论文)
首先,该过程在一组分子结构上训练生成模型,以学习构建满足相同结构分布并类似于所学化学空间的分子的基本规则。然后,使用最初训练的生成式深度学习模型来预测大量(在数千到数百万范围内)新分子。对分子结构进行有效性检查,并根据结构过滤系统:例如,丢弃重复或断开连接的系统。对于结构生成,研究人员使用生成式自回归深度神经网络 G-SchNet。与大多数生成模型相比,G-SchNet 能够预测分子的结构组成和三维构象,可以作为电子结构计算和电子结构深度学习模型的输入。
深度神经网络 SchNet + H 促进了分子特性的筛选,实现了高计算效率。
通过在功能有机分子的 OE62 数据集上训练 G-SchNet 来演示所提出的工作流程。OE62 数据由具有高度化学和结构多样性的分子组成。
在这里,证明通过迭代地将 G-SchNet 偏向所需的属性范围。可以设计出位于原始训练数据集所代表的属性分布之外的 ΔE、IP 和 EA 值的分子。
用于以前未见过分子的(多)特性驱动设计
所提出的方法构成了一个高效的工作流程,用于以前未见过的分子的(多)特性驱动设计。
该模型的局限性之一是,它需要在每个循环中预测和筛选几十万个分子,以获得足够多的分子数量,从而在筛选后可以使生成模型产生偏差。这一过程是有限制的,特别是当生成结构的化学多样性很小时,如果分子被筛选为两种以上的属性,则可能成为计算瓶颈。这种限制可以通过条件生成模型来解决,例如条件 G-SchNet,它可以通过在训练期间将感兴趣的属性作为标签来调节生成模型,以预测具有某些属性的分子。
生成可行分子的能力并不是 G-SchNet 独有的,该研究所提方法的新颖之处在于其高吞吐量能力。
此外,以要求的精度通过量子化学计算筛选 1,000 个生成的分子,将花费超过 500,000 个计算小时或大约 20,000 天。相比之下,在这项工作中,研究人员在几天内筛选了数十万个分子。因此,此处应用的机器学习模型的组合是一个明显的优势,可提供真正的高通量分子设计能力。
该方法预测具有超出初始训练数据集的电子特性的分子的能力将有助于从高通量药物发现到有机电子分子设计的一系列应用。
未来的工作将探索如何通过使用不同的神经网络架构来进一步提高该方法的性能。
参考内容:https://techxplore.com/news/2023-02-ai-futuristic-electronics-bendy-tvs.html

 友情链接
友情链接