








公众号/
编辑 | 萝卜皮

人工智能有可能在进化的规模上开启对蛋白质结构的洞察力。直到 2022 年才有可能将蛋白质结构预测扩展到 2 亿个编目蛋白质。对大规模基因测序实验揭示的呈指数增长的数十亿蛋白质序列的结构进行表征,需要在折叠速度上取得突破。
在这里,Meta AI 团队和纽约大学的研究人员展示了,使用大型语言模型从主序列直接推断结构,可以在高分辨率结构预测中实现一个数量级的加速。
利用语言模型在数百万序列中学习进化模式的洞察力,该团队训练了多达 15B 个参数的模型,这是迄今为止最大的蛋白质语言模型。随着语言模型的缩放,它们学习的信息能够以单个原子的分辨率预测蛋白质的三维结构。这导致预测速度比最先进的技术快 60 倍,同时保持分辨率和准确性。
在此基础上,研究人员提出了 ESM Metage-nomic Atlas。这是宏基因组蛋白质的首次大规模结构表征,具有超过 6.17 亿个结构。该图谱揭示了超过 2.25 亿个高置信度预测,其中数百万个预测的结构与实验确定的结构相比是新颖的,从而以前所未有的视角了解地球上一些最不为人知的蛋白质结构的广度和多样性。
该研究以「Evolutionary-scale prediction of atomic level protein structure with a language model」为题,于 2023 年 3 月 16 日发布在《Science》。

语言模型从进化入手研究蛋白质序列
进化规模的蛋白质序列包含生物结构和功能的图像。这是因为蛋白质的生物学特性限制了其序列的突变,这些突变是通过进化选择的,将结构和功能记录到进化模式中。在一个蛋白质家族中,可以从序列模式中推断出结构和功能。从经典方法开始,通过引入深度学习,直到目前最先进的技术,这种洞察力一直是计算结构预测进展的核心。
生物结构和功能反映在蛋白质序列模式中的想法,也激发了对进化尺度语言模型的新研究。从香农的文本熵模型开始,已经开发出越来越复杂的语言模型来适应文本的统计,最终导致现代大规模基于注意力的体系结构。在数百万种不同蛋白质的氨基酸序列上训练的语言模型,有可能学习所有这些蛋白质的模式。这个想法与从蛋白质序列推断的标准基础形成对比,后者是从总结相关蛋白质进化模式的多序列比对开始。
在人工智能中,文本的语言模型,尽管它们的训练目标很简单,例如填充缺失的单词或预测下一个单词,但显示出可以开发与文本的潜在含义相关的新兴能力。这些能力随着规模的变化而发展,随着计算、数据和参数数量的增加,能力会越来越强。包含数百至数千亿个参数的现代语言模型开发了诸如小样本语言翻译、常识推理和数学问题解决等能力,所有这些都没有明确的监督。这些观察提出了一种可能性,即在蛋白质序列上训练的语言模型可能会表现出一种平行的出现形式。
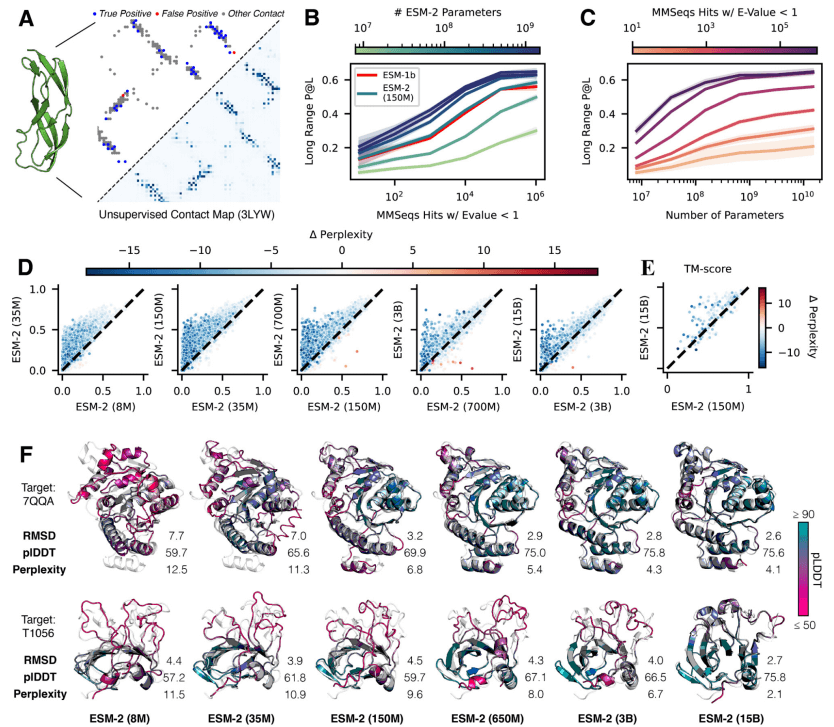
图:将语言模型扩展到 150 亿个参数时出现结构。(来源:论文)
研究人员假设,在整个进化过程中填补蛋白质序列中缺失的氨基酸的任务,将需要一个语言模型来学习一些关于在序列中创建模式的底层结构。随着语言模型的表示能力和训练中看到的蛋白质序列的多样性增加,科学家预计有关蛋白质序列生物学特性的深层信息可能会出现,因为这些特性会产生在序列中观察到的模式。
将参数扩展到 150 亿
为了研究这种涌现,研究人员将语言模型从 800 万个参数扩展到 150 亿个参数。科学家发现原子分辨率结构预测在参数尺度的四个数量级上出现并在语言模型中继续改进。语言模型对蛋白质序列的理解(困惑度)与结构预测的准确性之间的强相关性揭示了语言建模与结构学习之间的密切联系。
Meta AI 团队和纽约大学的研究人员展示了,语言模型可以直接从序列中进行快速的端到端原子分辨率结构预测。新方法利用语言模型捕获的进化模式,来产生准确的原子级预测。这消除了当前最先进的结构预测管道的昂贵方面,消除了对多序列比对的需要,同时大大简化了用于推理的神经架构。这导致仅推理前向传递的速度提高了 60 倍,同时还完全删除了相关蛋白质的搜索过程,使用 AlphaFold 和 RosettaFold 使用的高灵敏度管道可能需要 10 多分钟,即使使用新的灵敏度较低的快速管道,这也是计算成本的重要组成部分。在实践中,这意味着使用的最先进的预测管道的加速高达一到两个数量级。
这使得将结构预测扩展到宏基因组蛋白成为可能。在过去的十年中,人们努力通过宏基因组采样将蛋白质序列的知识扩展到地球上巨大的微生物自然多样性。这些努力导致蛋白质序列数据库的规模呈指数增长,现在包含数十亿种蛋白质。虽然最近已经完成了人类蛋白质组中约 20K 蛋白质和 Uniprot 约 200M 编目蛋白质的计算结构表征,但庞大的宏基因组蛋白质对结构表征提出了更大的挑战。宏基因组结构的范围和多样性是未知的,是生物学知识的前沿,也是医学和生物技术新发现的潜在来源。
该团队展示了宏基因组资源的第一个进化尺度结构特征,折叠了 MGnify90 中的几乎所有序列,超过 617M 蛋白质。他们能够在 2 周内在包含 2,000 个 GPU 的异构集群上完成此表征,展示了对更大数据库的可扩展性。对超过225M个结构进行了高置信预测,揭示并表征了宏基因组空间中远离现有知识的区域,绝大多数(76.8%)高置信预测与UniRef90至少有90%的序列同一性,数千万(12.6%)预测与实验确定的结构不匹配。这些结果第一次大规模地展示了宏基因组蛋白质结构的广泛性和多样性。
在蛋白质序列训练的语言模型中出现原子分辨率结构
该团队首先研究高分辨率蛋白质结构的出现。他们在 800 万个参数到 150 亿个参数的范围内训练了一个新的 transformer 蛋白语言模型家族 ESM-2。相对于上一代模型 ESM-1b,ESM-2 引入了架构、训练参数方面的改进,并增加了计算资源和数据。由此产生的 ESM-2 模型系列在相当数量的参数下显著优于先前最先进的 ESM-1b(约 6.5 亿参数模型),并且在结构预测基准上它也优于其他最近的蛋白质语言模型。
ESM-2 语言模型使用掩码语言建模目标进行训练,该目标训练模型通过观察序列其余部分的上下文,来预测蛋白质序列中随机选择的氨基酸的身份。这会使模型学习氨基酸之间的依赖关系。尽管训练目标本身很简单且不受监督,但要在数百万进化不同的蛋白质序列上完成这项任务,需要模型在整个进化过程中内化序列模式。
研究人员希望这种训练也能使结构具体化,因为它与序列模式相关联。ESM-2 是根据 UniRef 蛋白质序列数据库中的序列进行训练的。在训练期间,从约 1.38 亿个 UniRef90 序列中对约 4300 万个 UniRef50 训练集群进行均匀加权的序列采样,以便在训练过程中模型看到约 6500 万个独特序列。
当将 ESM-2 的规模从 800 万个参数增加到 150 亿个参数时,研究人员观察到其蛋白质序列建模的保真度有了很大的提高。这种保真度可以使用困惑度来衡量,其范围从 1(完美模型)到 20(随机预测模型)。直观上,困惑度描述了模型为每个预测选择的氨基酸数量。
图:ESM-2 掩码语言建模训练曲线。(来源:论文)
这种训练还导致模型中出现结构。由于 ESM-2 的训练仅针对序列,因此任何关于结构发展的信息都必须是表示序列模式的结果。已知使用掩码语言建模训练的 Transformer 模型会开发与蛋白质的残基接触图相对应的注意力模式。
该团队研究了这种蛋白质结构的低分辨率图片是如何作为比例函数出现的。研究人员使用线性投影从语言模型的注意力模式中提取接触图。顶部 L(蛋白质的长度)预测接触的精度(长距离接触精度)测量注意模式与蛋白质结构的对应性。
注意力模式在 ESM-2 中发展,对应于三级结构,缩放导致对结构的理解有很大的改进。预测接触的准确性随训练集中进化相关序列的数量而变化。训练集中具有更多相关序列的蛋白质在模型规模方面具有更陡峭的学习轨迹。这意味着对具有高进化深度的序列的改进在较低的模型尺度下饱和,而对具有低进化深度的序列的改进随着模型规模的增加而继续。
对于单个蛋白质,经常可以观察到作为比例函数的接触预测准确性的非线性改进。语言建模目标与注意力图中折叠结构的具体化直接相关。
为了识别模型中的原子分辨率信息,研究人员使用等变 transformer 从语言模型的内部表示中投影出每个原子的空间坐标。该预测适合使用来自 PDB 的实验确定的蛋白质结构,并在 194 种 CAMEO 蛋白质和 51 种 CASP14 蛋白质上进行评估。TM-score 的范围从 0 到 1,衡量投影与真实结构相比的准确性,值为 0.5 对应于正确预测折叠的阈值。评估使用时间截止,确保用于测试的蛋白质与用于拟合投影的蛋白质分开。这使得可以测量原子级信息如何作为参数尺度的函数出现在表示中。
研究发现,可以从 ESM-2 语言模型的表示中预测原子分辨率结构预测。该投影的准确性随着语言模型的规模而提高。150 亿参数模型在 CAMEO 测试集上的 TM 分数达到 0.72,在 CASP14 测试集上达到 0.55,相对于 1.5 亿参数的 ESM-2 模型分别提高了 14% 和 17%。
在每次规模增加时,蛋白质子集的准确性都会发生很大变化。例如,当参数规模从 35M 增加到 150M 时,蛋白质 7QQA 的 RMSD 从 7.0 提高到 3.2,而当参数规模从 3B 增加到 15B 时,CASP 目标 T1056 的 RMSD 从 4.0 提高到 2.6。在这些跳跃前后,RMSD 的变化要小得多。
在所有模型中,验证困惑度与 CASP14 TM-score 之间的相关性为 -0.99,验证困惑度与 CAMEO TM-score 之间的相关性为 -1.00,表明对通过困惑度测量的序列的理解与原子分辨率结构预测之间存在很强的联系。此外,可以从注意力图中提取的结构的低分辨率图片与原子分辨率预测之间存在很强的相关性(远程接触精度和 CASP14 TM-score 之间为 0.96,远程接触精度和 CAMEO TM-score 之间为 0.99)。这些发现将语言建模的改进与低分辨率(接触图)和高分辨率(原子级)结构信息的增加联系起来。
使用语言模型加速准确的原子分辨率结构预测
语言模型极大地加速了最先进的高分辨率结构预测。语言模型内化了与结构相关的进化模式,消除了对外部进化数据库、多序列比对和模板的需求。研究人员发现 ESM-2 语言模型可以直接从主要蛋白质序列生成最先进的三维结构预测。这导致结构预测的速度提高超过一个数量级,同时保持高分辨率精度。
通过为 ESM-2 训练折叠头来开发 ESMFold,这是一个完全端到端的单序列结构预测器。在预测时,蛋白质序列被输入到 ESM-2。该序列通过语言模型的前馈层进行处理,并将模型的内部状态(表示)传递给折叠头。头部以一系列折叠块开始。每个折叠块在更新序列表示和成对表示之间交替。这些块的输出被传递到等变 transformer 结构模块,并在输出最终原子级结构和预测置信度之前执行三个循环步骤。
与当前最先进的结构预测模型相比,该架构实现了重大简化,后者通过跨 MSA 的行和列运行的注意力机制将多序列比对深度集成到神经网络架构中。该方法显著提高了预测速度。在单个 NVIDIA V100 GPU 上,ESMFold 在 14.2 秒内对具有 384 个残基的蛋白质进行预测,比单个 AlphaFold2 模型快 6 倍。在较短的序列上,改进最多可达 60 倍。使用已发布版本的 AlphaFold 和 RosettaFold 使用的高灵敏度协议,构建 MSA 所需的相关序列搜索过程可能需要 10 分钟以上;这可以减少到不到 1 分钟,尽管灵敏度会降低。

图:使用 ESMFold 进行单序列结构预测。(来源:论文)
该团队在约 25K 个簇上训练折叠头,该簇覆盖了来自 PDB 的总共约 325K 个通过实验确定的结构,并进一步增加了用 AlphaFold2 预测的约 12M 个结构的数据集。该模型使用与 AlphaFold 相同的损失进行训练。
为了评估结构预测的准确性,研究人员使用在 2020 年 5 月截止日期之前从训练数据中提取的测试集;因此,评估中使用的所有结构都从训练中剔除,并且评估代表了在常规使用中预期的性能,作为实验者选择用于表征的结构类型的预测模型。这也使得与 AlphaFold 和 RosettaFold 进行比较成为可能,因为这些模型也没有接受过 2020 年 5 月之后沉积的结构的训练。这里使用了两个测试集:CAMEO 测试集包含 194 个用于正在进行的 CAMEO 评估的结构;CASP14 测试集由 51 个公开发布的结构组成,这些结构因其难度而被选为一年两次的结构预测竞赛。
研究人员将这些评估集的结果与 AlphaFold2 和 RosettaFold 进行比较。ESMFold 在 CAMEO 上的平均 TM-score 为 0.83,在 CASP14 上的平均 TM-score 为 0.68。使用与 AlphaFold2 一起发布的搜索协议,包括 MSA 和模板,AlphaFold2 在 CAMEO 和 CASP14 上分别达到 0.88 和 0.85。
ESMFold 在 CAMEO 上与 RosettaFold 实现了竞争精度,平均 TM-score 为 0.82。当通过消融多序列比对在单个序列上评估 AlphaFold2 和 RosettaFold 时,性能大幅下降,远低于 ESMFold。
另外,这是一个人工设置,因为 AlphaFold2 尚未针对单个序列进行明确训练,但它最近在蛋白质设计中变得很重要,这些模型已与单序列输入一起用于从头蛋白质设计。
由于语言模型是 ESMFold 的关键组成部分,研究人员测试了语言模型对序列的理解差异与结构预测准确性变化的对应关系。ESMFold 在两个测试集上的性能与语言模型的困惑度密切相关。在 CAMEO 测试集上,语言模型的困惑度与预测结构和实验结构之间的 TM-score 的 Pearson 相关性为 -0.55;在 CASP14 上,相关性为 -0.67。困惑度与结构预测之间的关系表明,改进语言模型是提高单序列结构预测准确性的关键,这与缩放分析的观察结果一致。此外,这意味着语言模型对序列的困惑度可用于预测 ESMFold 结构预测的质量。
消融研究表明,语言模型表示对 ESMFold 性能至关重要。对于 8 块折叠块,CAMEO 测试集的性能为 0.74 LDDT(基线)。如果没有语言模型,这会大大降低到 0.58 LDDT。当完全移除折叠树干时(即仅使用语言模型和结构模块),性能会下降到 0.66 LDDT。其他消融:结构模块只有 1 个块,关闭回收,不使用 AlphaFold2 预测结构作为蒸馏目标,或者不使用三角更新,导致小的性能下降(LDDT 从 -0.01 到 -0.04 的变化)。

图:CAMEO 和 CASP14 上的 ESMFold 消融。(来源:论文)
ESMFold 提供最先进的结构预测准确性,在超过一半的蛋白质上与 AlphaFold2 性能相匹配(< 0.05 LDDT 差异)。即使在一些大蛋白上也是如此——T1076 是一个 TM-score 为 0.98 且有 540 个残基的例子。ESMFold 和 AlphaFold 之间准确度较低的结构部分没有显著差异,这表明语言模型正在学习类似于 MSA 中包含的信息。并且,ESMFold 能够很好地预测同源和异源二聚体蛋白质复合物的成分。在与存放在 PDB 中的 2,978 个近期多聚体复合物的数据集上与 AlphaFold-Multimer 进行比较时,ESMFold 对 53.2% 的链对实现了相同的定性 DockQ 分类,尽管没有接受蛋白质复合物的训练。
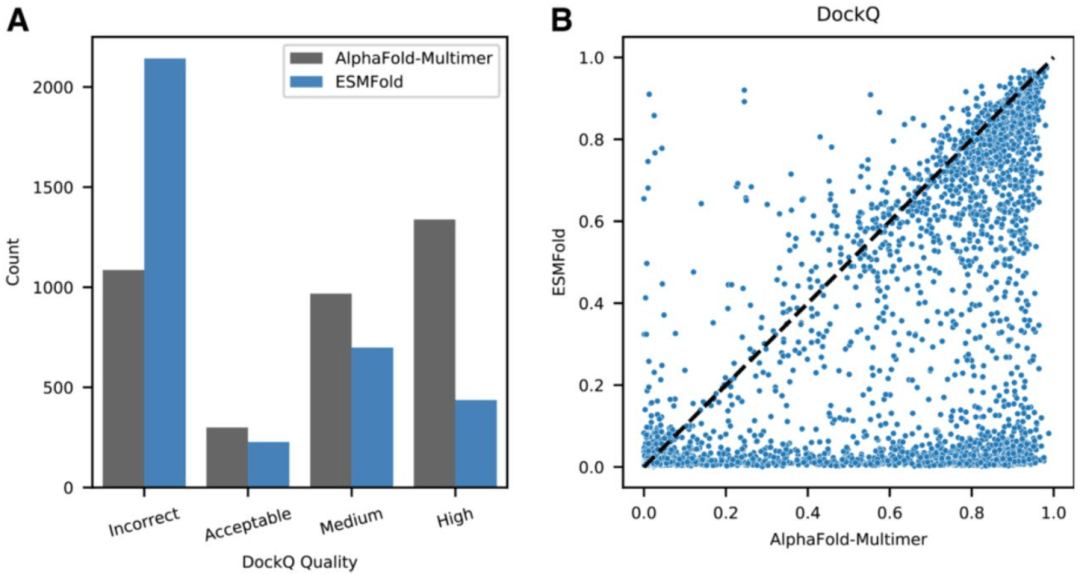
图:ESMFold 和 AlphaFold-Multimer 在 recent-PDB-multimers 数据集上的比较。(来源:论文) 信心是用准确度很好地校准的。ESMFold 以预测的 LDDT 形式报告置信度。这种置信度与预测的准确性密切相关,对于高置信度预测 (pLDDT > 0.7),准确性与 AlphaFold2 相当(ESMFold LDDT=0.83,AlphaFold2 LDDT=0.85 on CAMEO)。高置信度预测接近实验级精度。在 CAMEO 测试集上,ESMFold 预测的全原子 RMSD95 中位数(95% 残基覆盖率下的均方根偏差)为 1.91 Å,骨干 RMSD95 为 1.33 Å。当置信度非常高时 (pLDDT > 0.9),预测的全原子 RMSD95 中位数为 1.42 Å,骨架 RMSD95 为 0.94 Å。这意味着置信度可用于预测给定结构预测与真实结构匹配的可能性(要通过实验确定)。
宏基因组学的进化尺度结构表征
这种快速和高分辨率的结构预测能力使大型宏基因组序列资源的第一个全面结构表征成为可能。研究人员从 MGnify90 数据库中折叠了超过 6.17 亿个序列。这是长度为 20 到 1024 的整个序列,涵盖了 MGnify90 中所有序列的 99%。
总体而言,这种大规模表征产生了约 3.65 亿个具有良好置信度的预测(平均 pLDDT > 0.5 和 pTM > 0.5),对应于数据库的约 59%,以及约 2.25 亿个具有高置信度的预测(平均 pLDDT > 0.7 和 pTM > 0.7),对应于约 36% 的折叠结构。该团队能够在 2 周内在大约 2,000 个 GPU 的集群上完成预测。

图:映射宏基因组结构空间。(来源:论文)
结语
早期模型在许多任务上的表现甚至无法与简单的进化特征相媲美。对 ESM-1b 和 ProtTrans 等最先进的进化比例模型的分析表明,可以从表示中恢复低分辨率结构,即二级结构和接触图。
进化尺度模型也被证明可以执行突变效应的无监督预测,并且最近被用于最先进的应用,例如预测病毒进化的路径和基因变异的临床意义。几个大型模型现在可以作为开源使用。语言模型已经被研究用于骨干结构的端到端单序列预测。
快速准确的计算结构预测,有可能加速迈向一个可以理解基因测序实验中发现的所有蛋白质结构的时代。这有望对蛋白质的巨大自然多样性产生新的见解,其中大部分是在宏基因组测序中新发现的。
为此,该团队完成了宏基因组蛋白的首次大规模结构表征。这种表征揭示了数亿种以前未知的蛋白质的结构,与实验确定的结构相比,其中数百万种蛋白质是新的。
随着结构预测继续扩大到更多的蛋白质,模型的校准将成为一个关键因素,因为当预测的吞吐量受到限制时,预测的准确性和速度形成了可以生成的准确预测数量的联合边界。宏基因组图谱中非常高置信度的预测通常是可靠的,分辨率足以洞察类似于实验确定的结构,例如活性位点的生物化学;对于更多可以可靠地预测拓扑结构的蛋白质,可以通过远程结构关系获得对功能的洞察力,而这些关系无法通过序列检测到。
语言模型中原子级结构的出现揭示了蛋白质结构的高分辨率图片,该图片通过进化编码成跨越数百万蛋白质的序列模式,增加了无监督训练目标具体化蛋白质生物学深层信息的证据。ESM-2 是该团队多年来专注于生物特性出现的工作成果,并且是语言模型首次被证明可以捕获高分辨率的结构图片。目前的模型在参数、序列数据和原则上可以应用的计算方面远远没有达到规模限制。研究人员乐观地认为,随着继续扩大规模,将会有更多的出现。该团队的结果表明低深度蛋白质建模的改进指向了这个方向。
ESM-2 带来了速度的提高,实际上提高了一到两个数量级,这使得更多的序列能够准确预测原子级结构。在实际时间尺度内获得数亿个预测结构,有助于揭示对天然蛋白质的广度和多样性的新见解,并加速新蛋白质结构和功能的发现。
图谱资料:https://esmatlas.com
论文链接:https://www.biorxiv.org/content/10.1101/2022.07.20.500902v3.abstract

 友情链接
友情链接